Fenilketonurija je kongenitalna bolest. U većini nevezanih bolesnika, bolest se manifestira jednim stupnjem mentalne retardacije. Što je fenilketonurija? Čitati o tome u članku.
Sadržaj
 Fenilalanin se odnosi na neophodne aminokiseline (tvari koje idu u konstrukciju proteina u ljudskom tijelu), budući da životinjske tkanine nisu sposobne za sintezu. U ljudskom tijelu, gdje fenilalanin padne s hranom, proces njegove oksidacije je u tijeku pomoću specifične enzim fenilalanninske hidroksilaze do potpuno zamjenjive aminokiselinske kiseline - tirozin. Povreda (blokiranje) ove reakcije, uočeno kršenje sinteze fenilalananinhydroksilaze u jetri, dovodi do razvoja teške nasljedne bolesti - fenilketonurije.
Fenilalanin se odnosi na neophodne aminokiseline (tvari koje idu u konstrukciju proteina u ljudskom tijelu), budući da životinjske tkanine nisu sposobne za sintezu. U ljudskom tijelu, gdje fenilalanin padne s hranom, proces njegove oksidacije je u tijeku pomoću specifične enzim fenilalanninske hidroksilaze do potpuno zamjenjive aminokiselinske kiseline - tirozin. Povreda (blokiranje) ove reakcije, uočeno kršenje sinteze fenilalananinhydroksilaze u jetri, dovodi do razvoja teške nasljedne bolesti - fenilketonurije.
Tirozin koristi tijelo za sintezu hormona štitnjače. I derivat melanina, tirozina i fenilalanina, osigurava pigmentaciju (boju boje) kože, oko i kose.
Fenilketonurija se također naziva fenilalinanimija, fenilpirogradna oligofrenija. Bolest se odnosi na kongenitalne metaboličke poremećaje (metabolizam), karakteriziran je povećanjem razine fenilalanina u krvnoj plazmi i popraćena je mentalnom retardacijom. Točan uzrok mentalne retardacije u fenilketonuriji je nepoznat, ali to je posljedica biokemijskog defekta. Nasljedna priroda bolesti nalazi se u većini ljudi. Učestalost pojave FCU je 1 slučaj za 6000-10000 novorođenčadi.
Simptomi fenilketonurije
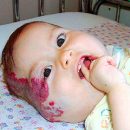
Klinički simptomi fenilketonurije u novorođenčadi su obično odsutni, tako da će se provesti laboratorijski probir (dijagnostika). U rijetkim slučajevima, letargija je zabilježena i poteškoća. U većini neprebilnih pacijenata, bolest se manifestira jednim stupnjem mentalne retardacije (češće izraženo), koji je glavni simptom fenilketonurije. Obično pacijenti s svjetlijom kožom, kosom i očima od njihovih zdravih rođaka. Neka djeca mogu imati promjene kože koje podsjećaju na dječje eCase.
Za bolesnike s fenilketonurije, karakterizirani su izobiljeni neurološki simptomi, refleksi se posebno mijenjaju. U starijoj djeci zabilježene su i veliki i mali epileptički napadaji, odstupanja na elektroencefalogramu zabilježene su u 75 - 90% slučajeva. Izraženo povećanje aktivnosti i psihoze razvija, često od pacijenata dolazi neugodno "miš" Miris uzrokovan prisutnošću feniloksične kiseline u urinu i znoju.
Dijagnoza fenilketonurije
Nakon što je novorođenče primljeno mlijeko (izvor fenilalanina) najmanje 48 sati, liječnici provode test probira na fenilketonuriji (ispitati kap krvi). U dobi od 4 do 6 tjedana, proizvod propadanja fenilalanina može se otkriti u urinu pacijenta. U ovom trenutku provodi se još jedan test skrininga na fenilketonuriji. Oštrica se provodi nakon završetka novorođenčeta, a djeca iz obitelji gdje postoji pacijent fenilketonuria, treba se redovito ponavljati tijekom prve godine života.
Liječenje fenilketonurije
Tretman je ograničiti ulazak fenilalanina s hranom kako bi se zadovoljila potreba za ovom neophodnom amino kiselinom, ali ne i više; Osigurati normalan rast i razvoj, ne dopuštajući akumulaciju u tijelu fenilalanina i konačnih proizvoda od njegove razmjene. Trajno praćenje je potrebno za razinu fenilalanina u djetetovoj krvi. U Sjedinjenim Američkim Državama zamijeniti mlijeko, proizvod Lofenalk se široko koristi, pun u svim aspektima koji sadrže malu količinu fenilalanina. Dopušteno je koristiti niskokularne prirodne proizvode, kao što su voće, povrće, neke žitarice i t.D. Potreba za fenilalaninom je zadovoljna zbog potrošnje ograničenog broja prirodnog proteina i preostale količine fenilalanina u lofenalu. Trenutno postoje proizvodi koji su potpuno pročišćeni od fenilalanina. Proizvod fenil fri sadrži sve potrebne sastojke osim fenilalanina, ne sadrži masnoću, stoga je njegova energetska vrijednost niža od one od drugih proizvoda.
Liječenje liječnika treba držati od prvih dana života. Samo rano i dosljedno liječenje sprječava poraz središnjeg živčanog sustava i teške mentalne retardacije. Ako je tretman počeo nakon 2-3 godine života, može se ograničiti samo na izraženu povećanu aktivnost i olakšava tijek konvulzivnog sindroma.